- A+
肿瘤靶向治疗依赖于肿瘤基因突变检测,传统的基因突变检测方法有Sanger测序、qPCR和焦磷酸测序等,通常对单个基因或者几个基因的热点突变或部分外显子区域进行检测,如果对多个基因进行检测,则需要更多的样本量、更长的检测时间以及更大的工作量。
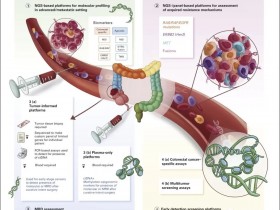
目前,临床分子实验室绝大多数都在进行基于NGS平台的肿瘤用药基因突变检测,能够同时对几十、几百甚至几万个基因进行热点、全外显子或全基因组的检测,而且需要的样本量并不增加,并且随着测序成本的不断下降,其在通量、效率方面的优势将会越来越明显,But,与此同时,对基因突变检测结果的解读和对报告形式制定的统一标准将显得非常重要。
依小编拙见,肿瘤基因突变检测将会是下一个超越NIPT的基因突变检测领域的爆发点,虽然目前很多公司都是亏本在做,不过从2017年FDA批准了两款NGS大panel产品及基因慧整理的2017年度的基因行业公司投融资来看(55家企业共融资了90亿元),2018年,投融资力度将会更加明显区分头部企业与尾部企业,加上国内的CFDA随时可能效仿FDA的政策,NGS产品也是很可能随时获批的,所以肿瘤基因突变检测行业随时可能会进入洗牌和重组的阶段。在如此情境之下,怎么才能提高一个基因突变检测公司的竞争力?除了价格、周期以外,就是服务的质量:基因突变检测报告的解读和批注(虽然我一直认为有能力的销售才是公司竞争力的根本,没有样本及市场,谈何生存与竞争?)。肿瘤基因突变检测目前急需一个统一的标准来解读和报告临床实验室发现的基因变异。
关于肿瘤基因突变检测的报告,小编觉得应该参考国外及国内的一些指南及专家共识:
建议参考2016年12月发表的文章:《Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer 》。
2015 年春开始,美国分子病理协会(AMP)召集多学科工作组,包括了分子病理学家,肿瘤临床医生,ACMG 的代表, ASCO 和 CAP 的专家等,对肿瘤及疑似肿瘤相关的序列变异检测建立检测标准并在行业达成共识。
该工作组开展了一项关于 NGS 技术和报告体系的调研,该调研在 AMP 团队成员的网站持续了大约 4 周。该调研最终收到 66 个关于 NGS 技术的结果,其中 44 个包含了 NGS 的报告内容。这些参与者报告了 NGS 技术在实体瘤和血液肿瘤上的检测结果,检测基因的数目从 1-10 个基因到大于 100 个基因。少数的参与者进行了外显子(12%)或全基因组(5%)的检测。所有的参与人员都表示可以检测 SNV突变; 95%的参与者表示可以检测小的 indel 突变;但是只有 35%的参与者报告可以检测 CNV,只有 37%的参与者表示可以检测基因融合。


根据临床影响将体细胞突变分成四级: I 类, 有较强的临床重要性的变异(等级A 和 B 证据); II 类, 有潜在的临床重要性的变异(等级 C 或 D 证据); III 类,临床意义未知的变异; IV 类, 良性或可能良性的变异感。兴趣可以下载原文看看。
2016年4月23日,CSCO与中国肿瘤驱动基因分析联盟(CAGC)发布《二代测序技术应用于临床肿瘤精准诊治的共识》,《共识》旨在为NGS技术应用于临床肿瘤驱动基因分析,提供相关指导性建议并规范临床实践。但是,到目前为止,该指南一直没有完全对大众公布。
所以,建议参考2017年3月《中华病理学杂志》第46卷第3期:《临床分子病理实验室二代基因测序检测专家共识》,此共识特色是基于病理评估的组织样本(FFPET、新鲜组织) 而形成的规范。

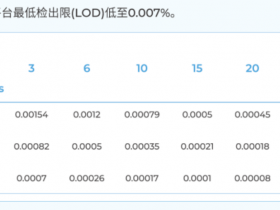
原始文件及比对结果文件的质量指标含义
NGS临床报告包含内容:基于高通量测序技术,临床实验室比较容易的获取更高通量的临床样本检测数据,不可避免会检测到意义未知的变异位点,在实际工作中会有一定的不确定性。但NGS的检测报告建议体现以下内容:
(1)检测名称,如XXX基因变异检测报告。
(2)患者基本信息,姓名,年龄,性别,住院号,送样医院科室及医师等。
(3)样本信息,病理号,取材部位,样本类型(FFPE、新鲜、血液等)、送检日期、报告日期等。
(4)病理信息,肿瘤组织类型、位置、TNM分期、细胞含量、肿瘤细胞比例、特殊说明(出血、坏死、酸脱钙处理等)。
(5)检测技术,包含所用基因panel、检测平台名称,分析软件版本号等。
(6)结果列表应包含,基因名称、变异在染色体位置、变异频率、cDNA的Genbank号(NM开头)及符合人类基因组变异协会(Human Genome Variation Society,HGVS)书写规范的突变类型、编码蛋白Genbank号(NP开头)及突变类型、杂合/纯合状态等。
(7)临床意义解读和批注:体细胞突变,报告各个肿瘤检测到的变异位点及临床意义。胚系突变,对于检测到的变异位点的致病性予以相应的临床解释。临床意义解读要客观平实的描述,在疾病相关性只描述既往研究中的疗效或预测,不能出现使用何种治疗手段或策略的语言。
(8)若检测失败,应阐述失败原因。
(9)最终报告应由检测者、报告医师或指定审核人联合签发。
在描述所检测出的基因变异时要遵循一定的原则和规范,推荐使用人类基因组变异协会命名指南。
对于肿瘤体细胞突变,根据突变的类型和已有的报道及指南,基因变异提倡分级的处理方式:
A级:美国食品药品管理局(FDA)或中国食品药品管理局(CFDA)批准的用药治疗靶点;写入中外诊疗指南有明确诊断/治疗/预后意义的变异。在报告注释该变异位点的临床诊断/治疗/预后意义的权威指南来源。
B级:尚未进入诊疗指南,但已经写入该领域的专家共识的变异位点。注释时要批注研究报道及专家共识的来源,明确其药物及其临床意义、正在开展的状态等信息。
C级:FDA或CFDA批准用于其他肿瘤可预测疗效的基因变异,或者正在进行中的临床试验变异位点。注释时要批注用于其他肿瘤的权威指南,研究文献及临床试验正在开展的状态等信息。
D级:处于学术争议或临床意义不明确的基因变异。同一实验室应该有统一的政策用来应对检测过程中出现的临床意义不明变异情况。
对于以上几种情况在报告的时候注意客观平实地描述检测的结果,在病理报告中不能出现建议使用何种治疗手段或策略的语言。
由于通量的增加和人种差异,临床肿瘤样本可能发现新的变异位点。实验室必须制定相关政策方案用来应对检测过程中出现的临床意义不明变异情况。政策可以是一发现变异就报告,但附上说明和意义。也可以是不报道这些发现或只报道小部分变异结果,并附上说明和参考文献及数据库。但是在报告的备注里一定要声明本实验室的报告规则。