- A+
Anti-PD药物的出现和临床应用,是肿瘤治疗的革命性进步,商业或临床均取得了巨大的成功。肿瘤具有自我演化能力,其复杂性和异质性导致针对癌症治疗的成功大多只是暂时的和局部的。时至2022年中,抗肿瘤治疗或许即将进入一个新时代。
这个新时代可能包括:探索肿瘤异质性以指导精准治疗、药物合成技术改变现有治疗模式(PROTAC、T-DXd、别构体抑制剂等)、基因编辑技术的商业化和临床应用。下面将通过这三个方向,结合最近发表的重磅文章进行学习和讨论。
探索肿瘤异质性以指导精准治疗
Anti-PD首次获批也不过在2014年,短短7-8年的时间,仅美国FDA就已经批准85个抗肿瘤适应症,超过2000个临床研究在评估33个同类产品,据说我国anti-PD产品更是超过100个。4月7日,Richard Pazdur在NEJM发表评论,参考如下。

高度内卷可能会带来诸多问题,1. 当对商业的考量超过创新时,重复、同质化的临床研究设计是对研究资源的巨大浪费。2. “知之为知之、不知为不知,是知也”。是时候停下来探索不知。3. 大军已占山头。
Anti-PD联合化疗已经成为肺癌、肝癌、胃癌、三阴乳腺癌等一众大瘤种的标准疗法,这种局面导致后面创新者都将面临重大挑战,如何鼓励后续创新需要科学指导、也需要监管部门的思考。换言之,假设SCLC和NSCLC一线都需要对比PD-L1联合化疗,怎么做?罗氏的TIGIT抗体Tiragolumab在这两个研究中都尝到苦果,不改变恐怕会有更多的创新被误伤。
这种观点的前提是,尽管一线anti-PD联合化疗的方案已逐渐标准化,但却是overestimate的,该联合方案并未解决所有问题。
例如,
1. 以NSCLC为例,PD-1为何对PD-L1阴性的患者也有效?
2. 为何临床特征(PD-L1水平、或MMR)相近,但治疗结局大相径庭?
3. 化疗理论上会抑制免疫反应,为何联合增效,而有些瘤种却不是?
4. 为什么Anti-CTLA-4在动物研究中发现其抑制Treg细胞和de novo T细胞,无法和PD-1发挥协同效应,但在人体研究中却发现了增效?
5. 为什么有些患者开始有效,但很快复发?6. Anti-PD药物仅对25%的实体肿瘤和40-60%的特定淋巴瘤有效,例如胰腺癌等肿瘤却尚未观察到疗效,为何有效又为何无效呢?
4月22日,作为《科学》杂志封面论文,刊登Ira Mellman的最新研究结果。细胞毒性T细胞和自然杀伤细胞在发挥抗肿瘤作用时,穿孔素和颗粒酶是关键要素。穿孔素负责在细胞膜上打孔,允许颗粒酶进入细胞诱导凋亡。然而,穿孔素打孔后,目标细胞中可能会出现一种蛋白(Endosomal sorting complexes required for transport,ESCRT)负责修复细胞破损,从而避免颗粒酶进入,发挥保护细胞(肿瘤的免疫逃避机制)功能。
为了进一步验证这一假设,通过CRISPR技术敲除细胞中编码ESCRT蛋白的基因,相比对照细胞,ESCRT受抑制的癌细胞可以被T细胞更有效的杀灭。从理论上来讲,开发一种药物抑制ESCRT蛋白,可能起到增强PD-1或CAR-T等作用。但实际上目前不可行,因为ESCRT蛋白参与正常细胞的分裂过程,除非找到只参与肿瘤免疫逃逸的那个特异性蛋白。

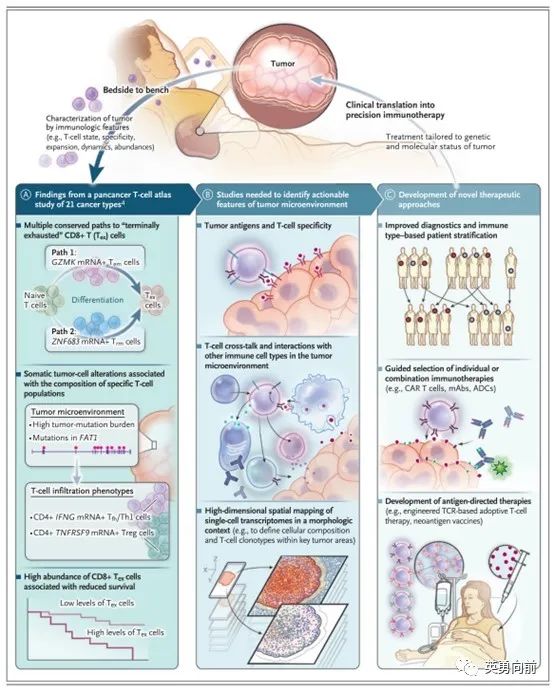
关于IO治疗,对肿瘤微环境的探讨尤为重要。6月14日,陈列平教授在NRDD发表重磅文章评述该问题,并给出建议,特别推荐仔细阅读和学习。

适应性免疫耐受(AIR)用于描述肿瘤微环境(TME)的不同状态,根据PD-L1和肿瘤浸润淋巴细胞(TIL)分为四种:PDL1-/TIL-、PDL1+/TIL+ 、PDL1-/TIL+ 、PDL1+/TIL-。
II型描述的是肿瘤微环境中PD-L1+且存在TIL,这种TME暗示肿瘤免疫逃逸是由于PD-1通路导致,因此Anti-PD药物应该有效。
I型则描述了一种TME中即不存在PD-L1也没有TIL,暗示肿瘤的免疫逃逸与PD-1通路是无关的,此类药物也不大可能有效。作为对应,可以考虑诸多策略,例如放化疗刺激肿瘤抗原的释放改变I型状态、或者使用适应性细胞疗法改变局部的肿瘤微环境,增强Anti-PD药物的疗效。
站在药物研发角度来看,有如下几点考虑。1. 如何对下述四种TME进行诊断的标准化,即CDx怎么开发?2. 仍然需要大样本的回顾性资料验证这一模型,并在不同疾病中前瞻性探索,通过IIT模式似乎可行性更高。
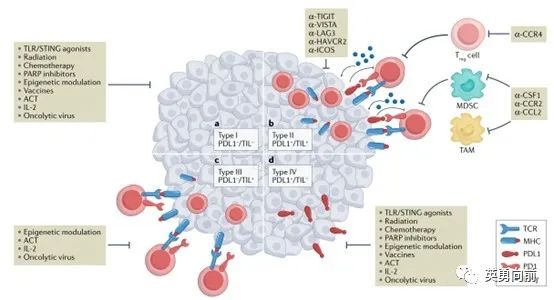
肿瘤微环境中,肿瘤细胞表面PD-L1和环境中的TIL对治疗的选择和预后起到关键作用。然而,TIL并非定性概念,不同肿瘤中TIL的功能和表型仍然不是很清楚。例如,TIL中包括一类终末期耗竭型CD8+的T细胞,此类细胞尽管是TIL,但实际是缺乏活性的。
为此,推荐一篇3月10日发表在NEJM的短文,值得学习。

利用单细胞 RNA 测序技术(Single cell RNA sequencing,scRNA-seq),一种在单细胞水平上利用 RNA 测序对特定细胞群体进行基因表达谱定量的高通量实验技术。作者描绘了一种bedside to bench的治疗场景,依据患者TME的状态选择更有针对性的肿瘤免疫方案。
通过活检获得肿瘤标本,检测TME的免疫状态、分析该状态下可能的不同情境、个体化治疗患者。在此过程中,需要不断标准化诊断技术、AI辅助分析TME、指导临床进行患者按免疫状态分组治疗。
换句话说,既然分子靶向可以如此细分,为何免疫治疗至今只有PD-L1和MSI-H/dMMR作为指导,况且还是模糊的。如果KRAS G12C没有突变,就绝不会使用KRAS抑制剂,但PD-L1阴性也可以使用PD-1抑制剂,这显然是有缺陷的。
对肿瘤异质性探索当然不仅局限于晚期癌症,也包括在局部晚期等非转移性癌症中。与ASCO同步发表在NEJM的一项IIT研究,12例接受dostarlimab单药(3周一次共6个月)治疗的局部晚期直肠癌(MMR)患者,至少随访6个月,全部获得临床CR且疗效持久,包括PET-CT、MRI、肠镜、活检病理全部均未发现肿瘤。
分析12例患者,所有人PD-L1和TIL均为阳性,也就是上面陈列平教授描述的Type II的AIR。
研究在ASCO上引起轰动,对于局部晚期MMR患者,单药PD-1可以临床治愈,且避免放化疗和手术,以及后者对患者生存质量不可逆的严重影响,同时把“创新往前”也是最近FDA的指导精神所在,不要局限于IV期。

6月23日,与该文章发表的同一天,Dr Hanna K. Sanoff在NEJM发表对该研究的短评。在高度肯定研究的同时也提出几点疑问,
1. 临床CR并非可靠的替代终点,大约20-30%患者会出现复发,且KEYNOTE-177研究中pembrolizumab单药治疗后仅55%患者在12个月能维持PFS。因此,dostarlimab获得的临床CR不等于治愈。
2. 研究仅入组单中心12例患者,无法反应此类患者的异质性问题,这种异质性包括治疗中心、合并症、肿瘤负荷、年龄等,在未完全明确之前,谨慎推进到临床实践中。

药物合成技术改变现有治疗模式
本段将学习并讨论近期热门进展,包括KRAS抑制剂、T cell engager(TCE)、ADC、PROTAC、迭代TKI等。
将无法成药的靶点成药化,是癌症治疗的主要方向之一。2021年5月FDA批准Sotorasib标志着针对KRAS突变的初步成功。说是初步,是因为G12C仅发生在4-14%的NSCLC,以及2-3%的消化道肿瘤中;说是成功,是因为Sotorasib作为别构体抑制剂,已成功驯服经典的不可成药KRAS。
Adagrasib是即将获批的第二个进入临床应用的Kras G12C抑制剂,临床I/II期关键研究数据发表在6月2日的NEJM上,入组116例患者,ORR为42.9%,中位PFS和OS分别为6.5和12.6个月,针对颅内转移的ORR为33.3%,报道的3-4级AE为44.8%,绝大多数为3级,治疗终止率为6.9%。
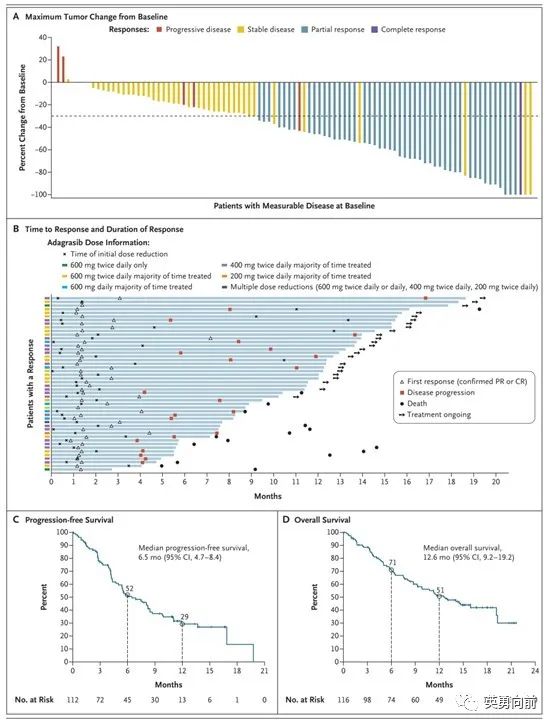

关于剂量探索问题,Sotorasib在获批后首次被FDA要求提供低剂量的数据,960mg QD的下一个低剂量为240mg QD,并需要在明年初递交给FDA审阅。
对剂量探索的担忧是最近FDA反复提及的问题,4月14日,FDA在Lancet Oncol发文阐述FDA在批准PI3K类药物中存在的问题和反思:生存是最根本的安全性终点。

PI3K-AKT-mTOR通路尽管在肿瘤细胞中过度表达,但同时也参与正常细胞的生理功能。2014年全球第一个PI3K抑制剂Idelalisib获批之后,一系列的安全性问题逐渐浮出水面,而在后续的开发中,研究人员通过靶向PI3K不同亚型,提高用药的耐受性和安全性。
安全性方面,此类药物较为常见的毒性反应为严重或致命性感染、中性粒细胞缺乏、免疫介导的毒性,严重AE的报告率可达到65%-85%。
这些AE对疾病本身是一种不良的循环起点,用药后的粒缺导致感染、感染导致停止治疗时间过长、过长的治疗中断导致疾病快速进展后死亡。
大多数关键研究的终点为ORR或PFS,除去PFS中包含的生存信息,临床研究数据体现肿瘤对药物的即时治疗反应,对长期的生存数据难以获得。
2022年4月21日,FDA召开ODAC会议,以16票同意1票弃权的结果要求:FDA对于PI3K类药物在血液肿瘤中的开发,考虑到药物的治疗窗较窄(有效 vs. 毒性)以及靶点本身的潜在风险,需要随机对照研究支持监管的批准考量。
尽管目前只是对PI3K治疗血液肿瘤提出的要求,但FDA在本文中明确指出的三类要求:
1. 对剂量探索提出更高要求,从单纯以耐受性为立足点的剂量爬坡原则,提高到结合早期疗效、安全性以及PDPK等指标的综合性数据。
2. 提倡早期随机对照研究,弱化单臂研究ORR的临床批准。
3. 注重对生存数据的获得。
关于对剂量探索的要求,FDA尚未发布指导规范,但大家可以参考6月20号CDE发布《单臂临床试验用于支持抗肿瘤药上市申请的适用性技术指导原则》,CDE给了具体的建议。6月28日,相应的论文发表在中华肿瘤杂志。

除常规要求外,值得关注的几点:
1. 对联合用药的考量,原则上联合治疗的关键研究不接受单臂试验设计。但是,“如果联合用药的有效性非常突出,且申请人可以提供充分、可靠的析因分析时,申请人也可以与监管机构沟通联合用药采用 SAT 支持上市的可行性。”
2. 对于纳入单臂研究的患者,务必符合“无有效治疗手段的肿瘤患者”,在IND、pre-IND、pre-NDA等阶段需要和监管部门沟通,并且在NDA阶段,监管部门将“逐例地梳理受试者既往治疗史,以确保符合条件”。
3. 如果肿瘤发生的驱动因素清晰,所开发药物针对该因素,且作用机制明确,可以考虑在单臂研究中进行不限瘤种的肿瘤开发。
4. 对罕见肿瘤的定义可以扩展到大瘤种中的罕见突变、治疗后复发难治、不限瘤种等情况,对罕见肿瘤的药物开发有可能不进行随机对照,凭单臂研究获批。
KRAS G12C抑制剂的成功是不可成药靶点的初步胜利,类似胰腺癌这种超过90%存在KRAS突变的患者,如何找到G12D、V等药物对治疗预后的改善至关重要。因此,以PROTAC技术为代表的蛋白降解为人们提供了想象空间。
PROTAC为蛋白降解靶向联合体,这一概念是2001年由Dr Crews提出,利用体内天然存在的蛋白质清理系统(泛素-蛋白酶体),降低蛋白水平治疗疾病。
PROTAC是一种异双功能分子,分别连结靶蛋白(目标蛋白质)的配体和E3连接酶(泛素化)的配体,通俗来讲:PROTAC把拆迁队(泛素)叫来,把房子(蛋白质)用挖掘机(蛋白酶体)给拆了。
PROTAC、分子胶等尚在临床研究中,分享3月21日发表在NRDD上的一篇综述。

单克隆双特异性抗体是新药开发的一大热门,目前批准的产品包括blincyto、hemlibra、amivantamab和vabysmo。
6月8日,罗氏的CD20/CD3双抗Lunsumio(Mosunetuzumab)获得了欧盟委员会的有条件上市批准,用于治疗先前至少接受过两次系统治疗的复发性/难治性 (R/R) 滤泡性淋巴瘤 (FL) 成年患者。
值得一提的是,这是全球第6款获批上市的双抗药物,也是全球首款上市的CD20/CD3双抗。对于CD3为靶向的TCE,如何管理好CRS和感染是重要课题。

抗体-药物偶联物(ADC)是新药开发的热点之一。Mylotarg是2000年5月17日全球第一个获批上市的ADC类产品,由于毒性原因辉瑞于2010年撤回上市申请,在三项临床研究支持下,FDA于2017年9月1日又重新批准其上市治疗CD33+的AML。
Mylotarg和SGN-CD33A均出现过较明显的肝脏毒性,考虑到CD33并非表达在肝细胞表面分子,因此肝脏毒性可能与ADC药物的铰链化疗药物有关。
随机药物合成技术的不断进步,以第一三共开发的Trastuzumab Deruxtecan(T-DXd)和罗氏开发的Polatuzumab Vedotin(POLIVY)为代表的ADC类药物从医学上证明了此类药物巨大的潜在价值。
1月27日,NEJM刊发POLARIX的阳性三期数据,用于一线DLBCL的治疗。对于一线DLBCL来说,过去20年里大部分的注册三期研究均以失败告终,来那度胺、硼替佐米、依鲁替尼等等。

POLARIX研究对比pola-R-CHP和R-CHOP治疗一线DLBCL,随机双盲1:1入组879例患者,主要终点为PFS。IPI 2分两组均为38%,GCB分别占55.8%和49.7%,ABC为30.9%和35.2%。研究中位随访28.2个月,主要终点研究者评估的2年PFS分别为76.7%和70.2%(HR 0.73,P=0.02),2年OS分别为88.7%和88.6%(HR 0.94,P=0.75),安全性特征相似。
几点看法:
1. 成功很难得,为一线DLBCL增加了新的治疗选择,但想立即改变标准治疗还是很困难的;
2. 相比一个有20年使用历史的R-CHOP来说,增加POLIVY会带来治疗费用的增加和潜在的安全性风险,而获益表现为统计学意义的2年PFS增加,但OS并没有任何差别;
3. 研究本身取得的统计学阳性,与样本量较大、IPI 2分比例、ABC不均衡分布等等因素也可能有关。
值得一提的是主要终点为研究者评估的PFS,相比中心评估其优点是减少了因不一致导致的过多删失,更贴近临床现实,而缺点是存在一定程度主观性并且需要即时的医学监察。难分伯仲,可以参考下文。

另一个明星ADC是第一三共的T-DXd,是HER2靶向抗体药物偶联物,由人源化抗HER2单克隆抗体、可裂解的四肽链接键和拓扑异构酶Ⅰ抑制剂喜树碱衍生物(DX-8951 derivative,DXd)进行偶联而成。
T-DXd的药物设计将靶向药的精准和化疗药的高效进行了优化组合,使其具有更高的药载比、特殊的偶联结构和旁观者杀伤效应,降低了药物毒副作用,还增强了杀伤肿瘤的能力。
T-DXd在其系列研究中取得了诸多成功:
DESTINY-Breast 01:是一项全球多中心、开放Ⅱ期试验,评估T-DXd用于治疗T-DM1耐药/难治性的HER2阳性不可切除和/或转移性乳腺癌患者的安全性与疗效,研究主要终点ORR为60.9%,借此研究获得FDA批准。
DESTINY-Breast 02:作为01研究的确证性试验,对比600例T-DM1经治的HER2阳性不可切除和(或)转移性乳腺癌患者,探索T-DXd(5.4 mg/kg,q3w)较研究者选择方案(曲妥珠单抗+卡培他滨或拉帕替尼+卡培他滨)的差异。主要研究终点为PFS,次要研究终点为总生存期(OS)。
DESTINY-Breast 03:是首次头对头三期研究,是全球首个在HER2阳性晚期乳腺癌二线治疗中挑战T-DM1的Ⅲ期临床试验。研究共纳入了524例患者,T-DXd相比T-DM1,降低72%的疾病进展或死亡风险(HR 0.2840,P=7.8×10-22),统计学差异和临床意义都相当显著。DESTINY-Breast 03研究确立了T-DXd在HER2阳性晚期乳腺癌中的疗效。
DESTINY-Breast 04:ASCO 2022中的重磅研究,研究取得PFS和OS双阳性结果,标志着T-DXd治疗获益人群将从HER2阳性拓展至HER2低表达(55%的乳腺癌患者)患者,乃至为整个晚期乳腺癌的临床实践带来颠覆性变革。以往这类患者会被归类为HR+/HER2-或三阴乳腺癌人群肿瘤,对传统HER2治疗是无效的。

T-DXd除乳腺癌外,在一系列涉及HER2表达的肿瘤中均有所探索,包括NSCLC和胃癌等。DESTINY-Lung01研究结果发表在1月20日的NEJM,共91例患者入组,中位随访13.1个月,ORR为55%(44-65%),中位PFS为8.2个月,OS为17.8个月。期待进一步研究结果。

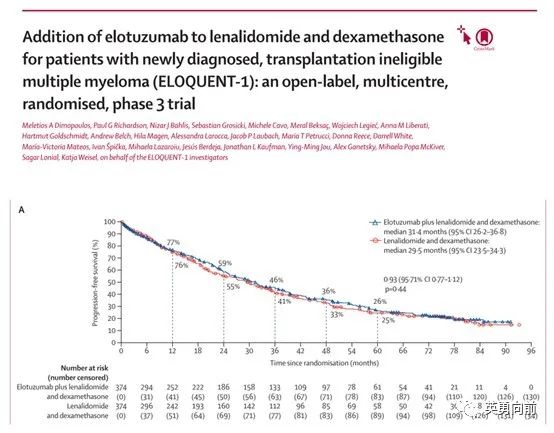
再介绍个曾经多多少少参与过的三期研究结果。ELOQUENT-1为探索Elotuzumab联合Rd方案治疗不能耐受移植的NDMM的三期研究。
研究从2011年8月入组至2014年6月19日,1:1随机748例患者接受ERd vs. Rd,中位年龄73岁,中位随访65.3个月,中位PFS分别为31.4个月和29.5个月(HR 0.93)。
相比daratumumab类似的三期MAIA研究,入组患者人数和基线很接近,PFS分别为未达到和34.4个月(HR 0.53,P<0.0001),60个月的PFS率分别为52.5%和28.7%,60个月的OS率为66.3%和53.1%(HR 0.68,P=0.0013)。
相比上述两个研究,对照组Rd的PFS相差不大,但治疗组的PFS相差是肉眼可见。几点感受,1. Elotuzumab靶向SLAMF-7(以前叫CS-1),主要表达在骨髓瘤细胞、NK细胞和一些免疫细胞表面,正常细胞表面不表达。
相比CD38,SLAMF-7是个更好的目标靶点。还记得2013年冬天,elotuzumab的2期数据在ASH前某酒店公布,我也有幸在场,满怀希望非常乐观。
一个体会是,机制再明确,也都要到大规模临床研究中去证伪,在这之前,保持谨慎乐观。
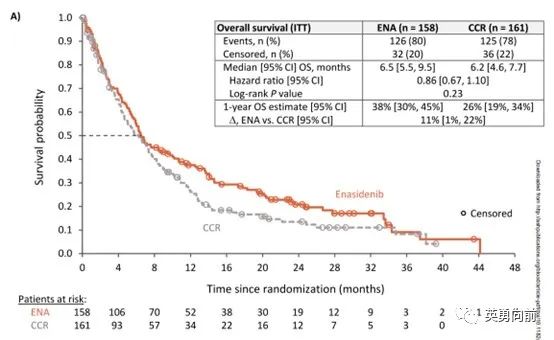
另外一个三期是enasidenib的三期,结果发表在6月17日的Blood杂志,主要终点OS分别为6.5和6.2个月。
几点体会,
1. 除非对治疗药物特别有把握,谨慎把医生选择作为对照组,增加过多不确定性,样本量的设定缺少前期数据支撑;
2. 以OS为终点的研究要慎重考虑不设盲,开放研究会增加患者退组意愿且后续抗肿瘤治疗肯定会影响OS结果,哪怕双终点也好过单一终点;
3. 研究过程中,过多的删失需要格外注意。
基因编辑技术的商业化和临床应用
以CD-19和BCMA作为靶点的自体CAR-T已经进入商业化阶段,其优势在于较好的疗效,局限在于高昂的生产成本、平均2-3周的生产周期、以CRS和神经毒性为代表的毒性反应等。尽管如此,细胞基因疗法取得的突破性进展是毫无疑问的。
自体CAR-T的开发策略之一是往前一步。
2月17日,ZUMA-7研究结果发表在NEJM,评估CAR-T对比标准治疗二线大B细胞淋巴瘤的临床三期研究。基于该研究结果,FDA于2022年4月15日批准YESCARTA治疗二线大B细胞淋巴瘤。
另外,6月25日,FDA也批准BMS开发的CD-19-CAR-T产品BREYANZI用于二线LBCL成人患者。
ZUMA-7研究1:1随机359例患者,主要终点EFS分别为8.3 vs. 2.0个月,CR率分别为65%和32%,2年OS分别为61% vs. 52%,CAR-T的3级CRS为6%,神经毒性21%。

1. 研究入组的人群包括DLBCL和高级别B细胞淋巴瘤,且74%的患者为原发难治、26%为12个月内复发。
2. 在CAR-T组中,36%患者接受了单纯激素桥接治疗,好在细胞制备的中位时间仅13天;而对照组94%的患者接受铂类药物为核心的治疗,36%接受了自体干细胞移植。
3. 研究主要终点选择了EFS,定义为从随机到疾病进展、开始新的淋巴瘤治疗、死亡、或随机后150天之内SD为最佳反应。EFS可以被监管部门接受,可以借鉴参考。
4. 在CAR-T的安全性方面,发热几乎都会出现,对血象的抑制也比较明显。尽管三级CRS仅6%但92%的患者均有CRS发生,主要表现为发热、低血压、窦速、缺氧和头疼等;60%的患者出现神经系统症状,主要表现为震颤、意识模糊、失语、脑病、感觉异常等。
Axi-Cel作为商业化的CAR-T产品,疗效比较明确但由于安全性问题会限制使用的人群,目前只能作为预后很差的患者的治疗选择。想再进一步估计还要进一步优化耐受性,改善可及性,缩短制备周期。
ZUMA-12研究也在4月发表在nature medicine上,这是一项临床二期研究,作为补充的一线治疗(R-CHOP或DA-EPOCH-R之后)高风险大B细胞淋巴瘤患者。
40例患者入组,在主要终点疗效可评估人群中(N=37),CR为78%,ORR为89%,3级及以上的CRS发生率为8%、神经毒性23%。CAR-T扩增的中位时间为8天。

自体CAR-T的应用也仅限于血液肿瘤中,细胞基因疗法作为商业化的产品,必须要考虑的问题包括:
1. 如何大幅降低价格?
2. 如何用于实体肿瘤和其他严重疾病?
3. 有什么方法进一步提高疗效和安全性?带着这些问题,来看看这些文章。
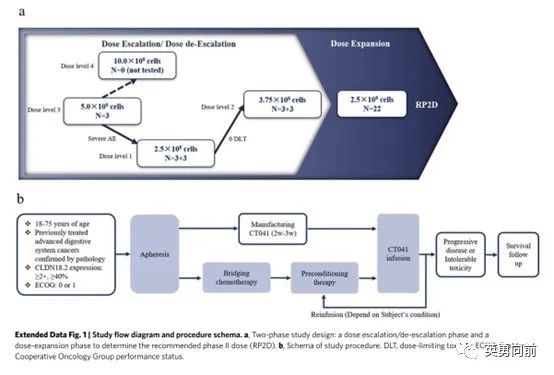
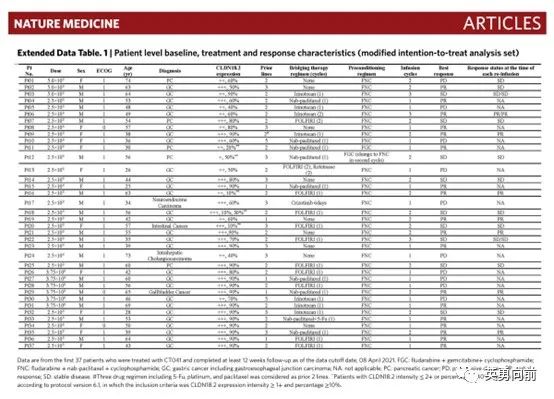
5月9日,沈琳教授牵头在Nature Medicine发表一篇Claudin 18.2-CAR-T(CT041)治疗复发晚期胃肠道肿瘤的I期研究。研究分为剂量爬坡(3+3,共18例)和扩展(22例)。
CT041共分计划三个剂量组:2.5*108/L(28例)、3.75*108/L(6例)、5.0*108/L(3例)。自2019年3月至2021年4月,74例患者签署ICF,49例患者接受回输,37例被纳入本次分析。

中位至回输时间为27天(22-187天),75.5%的患者接受了至少一个周期的桥接化疗,方案包括FOLFIRI(39.3%)、白蛋白紫杉醇(32.1%)或伊立替康(25%),桥接后至少15天会进行影响评估,仅5.5%-9.7%的患者出现肿瘤缩小。
35例患者接受氟达拉滨和环磷酰胺联合低剂量白蛋白紫杉醇(FNC)方案作为预处理,15例患者接受了二次细胞治疗。低剂量白紫据说可以降低免疫负调因子。
94.6%的患者出现1-2级CRS,无3级和以上CRS,27例患者接受anti-IL-6治疗,没有ICANS报道。疗效方面,所有37例患者的ORR为48.6%,CR为0,中位PFS为3.7个月。
文章提到,相比以往研究包括nivolumab、pembrolizumab、化疗和阿帕替尼,ORR大约在1.7%-13.3%,mPFS为1.6-2.6个月,OS不超过6个月。
相比这些数据,CT041的疗效有一些优势,但与CD19或BCMA作为靶点的疗效相差甚远,况且大部分患者接受了桥接化疗和预处理化疗,CT041的实际疗效多少有些混杂。
尽管如此,CT041还是证明了自体细胞疗法在实体肿瘤中可能有效,后续探索的方向可能:选出有效CAR-T细胞类型、探索联合和多次输注、探索现货供应型。
针对CAR-T治疗实体肿瘤差强人意的疗效,6月8日Nature杂志发表研究认为,使用IL-9受体进一步修饰CAR-T细胞,可以使后者增强抗肿瘤活性。

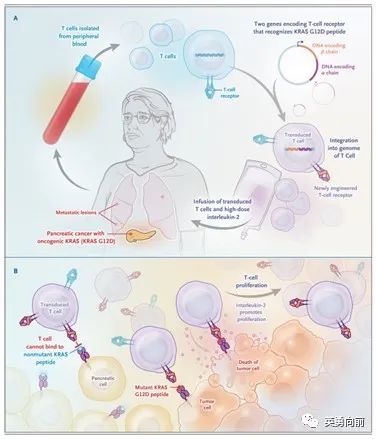
细胞基因治疗实体肿瘤,6月2日NEJM杂志发表了一个大胆的尝试,针对存在KRAS G12D突变的晚期胰腺癌患者。
单次回输16.2*109个自体CAR-T细胞,这些细胞表达异基因HLA-C*08:02限制性TCR并靶向KRAS G12D,回输6个月后,仍可以检测出2%的循环外周血T细胞。患者靶病灶均出现明显的缩小(62%-72%),获得部分缓解。

尽管只有个别患者数据,且HLA-C*08:02也只是占10%人群,但对于实体瘤这一大类来说,肿瘤抗原是不难找的,理论上这种细胞治疗方法有很多临床应用场景。
该细胞治疗的主要步骤如下:
1)选择HLA-C*08:02并分离患者自身外周血T淋巴细胞;
2)基因修饰T细胞TCR使其可以识别KRAS G12D肽段;
3)高剂量IL-2联合自体T细胞回输。
通用、现货、实体瘤是细胞疗法的目标之一,而CAR-NK是有潜力达成这一目前的产品类型。
CAR-NK细胞疗法也是近年来非常热门的开发领域,尽管尚无产品获批,但天然良好的安全性特征、可以反复多次给药、联合治疗的潜力、进入实体肿瘤、现货供应,以及在基因编辑技术背景下可以被不断优化的特点,CAR-NK的科学和商业潜力都是巨大的,目前来说科学重要性要大于商业的。
为进一步学习CAR-NK的一些特点,推荐3月21日发表在NRDD的一篇综述文章,以下将对此进行详细描述。

NK细胞是什么?
NK细胞和B细胞、T细胞一样是由同一类淋巴祖细胞分化而来,属于固有淋巴细胞,占10%的淋巴细胞。NK细胞可以在脾脏、肝脏、淋巴器官、胸腺、肠道、扁桃体、子宫等部位发育成熟,IL-15相关信号通路在淋巴祖细胞分化为CD3-CD56+的NK细胞过程中扮演重要角色。
NK细胞分为两大类:CD56brightCD16-(分布在组织中,可以产生细胞因子,缺乏IL-15情况下没有细胞毒性)以及CD56dimCD16+(分布在外周血中,发挥抗病毒和肿瘤作用)。
NK细胞在肿瘤微环境中的免疫细胞产生的趋化因子的引导下,进入肿瘤局部发挥作用。由于NK无需肿瘤抗原活化,因此对于MHC I类分子下调的肿瘤细胞(无法呈递肿瘤抗原),NK细胞仍然发挥抗肿瘤作用。
ADCC是NK细胞发挥抗肿瘤作用的另外一个重要机制,通过结合CD16发挥作用。除了上述两种针对MHC I类信号缺失和ADCC作用,间接增强CD8+T细胞的抗肿瘤活性也是一种方式。
影响NK细胞发挥抗肿瘤作用的TME包括IL-10、IDO、TGFb、Treg、缺氧、IL-37、MICA和MICB等,都可能作用联合super NK细胞疗法的联合药物选择。
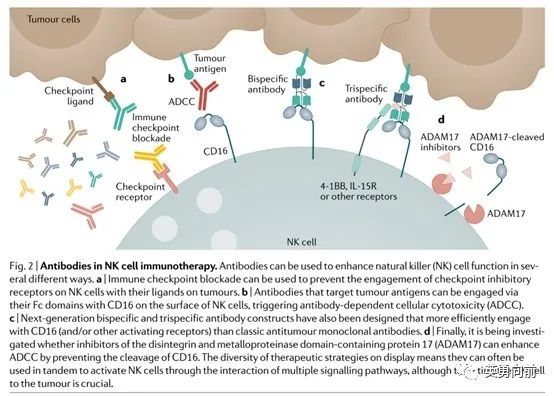
NK细胞疗法
把NK细胞作为一种商业化的产品正在进行中。首先解决NK细胞的来源问题,目前主要来源有四种:外周血(NK占10%的淋巴细胞)、脐带血(占30%淋巴细胞)、NK-92细胞系和诱导多能干细胞(iPSC)。
考虑到CMC的稳定质控和可行性,外周血和脐带血都存在很大的异质性,不适合作为商业化的NK细胞来源。NK-92是肿瘤细胞系,存在致癌风险,因此长期安全性仍不清晰。
iPSC是最常用的细胞来源,这是由日本科学家山中伸弥在2006年发明的技术,2012年获得诺贝尔奖。使用iPSC细胞可以保证NK细胞有无限增殖的可能,同时确保工艺稳定、质量参数一致,实现大规模较低成本的生产成为可能。
iPSC细胞在干细胞阶段,可以进行例如CRISPR基因编辑,使其表达目标分子。
NK细胞疗法走在前面的公司包括NKarta和FATE,在淋巴瘤和AML中见到初步的疗效,商业化还需要较长一段路要走。
可能需要考虑并解决的科学问题包括但不限于:
1. 如何使具有抗肿瘤活性的NK细胞定向到TME?
2. 在免疫抑制TME中,如何提高NK细胞抗肿瘤活性?3. 有哪些联合的可能性,例如,anti-PD、IgG1抗体(特别是西妥昔单抗和利妥昔单抗)、双/三抗。
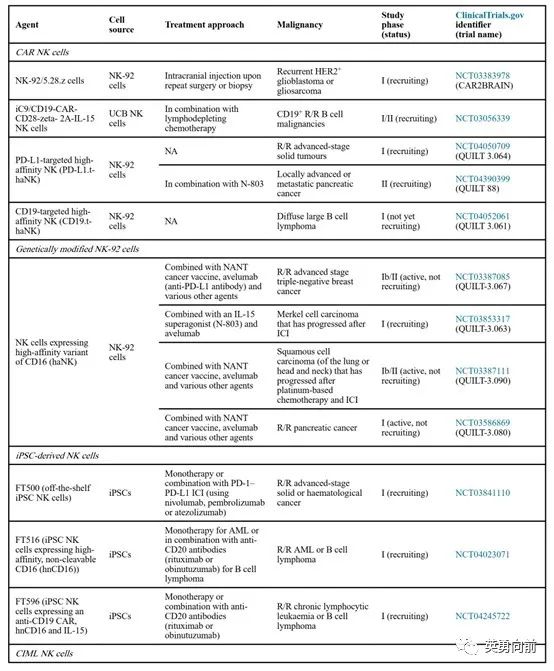
6月22日,Leukemia杂志刊发了(血液科)今年最重要的两篇必读文章,即WHO 2022分类标准(第5版WHO分类),包括髓系和淋巴系两大分类。
由于本文篇幅和有限的个人能力,只能简述部分内容。值得一提的是,复旦大学肿瘤医院李小秋教授作为共同作者参与了此全球指南的撰写。
髓系的几大变化:
1. CHIP和CCUS作为一种诊断,首次写进WHO分类标准中;
2. CML仅分为慢性期(CP)和急变期(BP),去除进展期(AP)分类;
3. MDS按照遗传学和形态学进行二分类诊断,其中的S代表的是neoplasms而不是以前的syndrome;
4. AML按照遗传学和分化情况分类诊断,且去除原始细胞20%的诊断线。

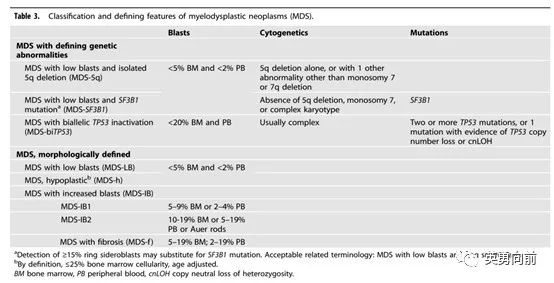
新版MDS分类相比上一版,
1. MDS-5q的诊断条件不变,检出SF3B1或TP53突变(非双等位基因)不影响MDS-5q的诊断,可以考虑来那度胺治疗。
2. MDS-SF3B1是新增独立诊断,SF3B1往往伴随RS细胞产生,预后较好,治疗与其他MDS可能不一样,例如使用luspatercept纠正贫血。
3. MDS-biTP53是新增独立诊断,TP53改变占7-11%的MDS患者,双等位基因占2/3患者(大约5-7%),此类患者预后差,因此对其治疗可以借鉴AML的诸多方案。
4. 按照形态学分类,根据患者原始细胞比例分为MDS-LB、MDS-IB1和2。
个人认为WHO 2022的MDS分类有助于临床治疗的选择,相比上一版(如MDS伴单系、RS、多系发育不全,MDS伴原始细胞增多等)要相对简单些。
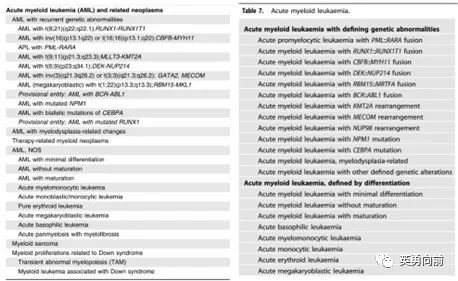
新版AML分类(下图右侧)相比上一版,
1. 取消了原来的AML NOS,代之简单明了的以遗传学和形态学(类似FAB的M0-M7)为标准的大的分类方法。
2. 遗传学分类标准中,不再强调20%原始细胞作为诊断AML的最低要求,但必须要明确该遗传学变化和形态学变化的因果关系。其中,BCR-ABL1和CEBPA还是需要采纳20%原始细胞作为诊断AML界值的。
3. 新增AML伴KMT2A、MECOM、NUP98,并且不要求20%,主要原因是以往这些患者会被诊断为MDS,而其临床特征与AML非常相似,需要更强烈的治疗。
个人认为WHO 2022的AML分类比较好记,特征性的遗传学变异可以支持诊断、FAB的诊断实践依然适用。另外,新分类侧重临床病理而非单纯病理诊断,需要结合临床、分子遗传学异常和病理学特征做出准确的诊断,并指导后续的治疗。
有一点不大理解,对于合并IDH1/2、FLT3 ITD/TKD突变的AML,假设原始细胞小于20%是不是也可以诊断AML而非MDS呢?还是因为遗传学与形态学的因果关系不够强么?
对于药物研发中的入组条件,还需要再谨慎一些。
淋巴系的几大变化,
1. 滤泡状淋巴瘤(FL)不再强制要求分级1-3a,新名称为经典型FL(占85%),而FL 3b改为滤泡大B细胞淋巴瘤(FLBL)。
2. DLBCL NOS不再区分GCB和ABC型,主要原因可能在于ROBUST研究没有成功,区分对指导治疗方案的选择价值不太大。

文末想推荐一篇4月27日发布在BioSpace网站上的短文,总结了成功的Biotech应具备的9种共同之处,供同行参考:
1. 多样化的人才团队:仅仅有最棒的科学家显然是不够的,作为一家企业所做的不仅仅是科学研究,更重要的是知晓开发一款产品直至商业化。
2. 经验丰富的董事会。
3.团队精神:透明、清晰的沟通和对人友善乃为公司的基石,CEO有责任去落实这些。
4. 战略性的财务考虑:每一轮融资的同时需要考虑下一轮的目的和时间表,而对于投资人的目标管理也非常关键,特别是投资早期生物技术公司的退出机制和预期。
5. 适应性:公司在不同阶段需要不同的资源。例如,科学家创始人需要根据公司发展阶段实时调整自己定位。
6. 患者为中心:应始终以解决患者的痛点为中心,如果一个疾病已经可以通过标准治疗得到非常好的控制,就没有必要花费巨额费用开发一个不确定性的东西。因此,生物技术公司必须要时刻保持和临床的沟通,起码要知道患者的需求在哪里。
7. 保持专注:成功的公司都是专注于某一领域,发挥其比较优势。尽管技术有非常宽广的适应性,但始终不能忘记专注。一个CMO不可能同时精通神经系统疾病、癌症和感染类疾病的。因此,专注于你擅长的,其他的找到合作方完成。
8. 清晰的生产计划:监管部门毫无疑问会质疑新的生产方法,因此,在开始阶段就应该和监管部门密切沟通,获得监管满意的生产方式。
9. 生态系统的建立:相比于传统的biotech生态系统理论,由于在线工作系统的畅通,对公司地理位置的要求不断弱化。但无论如何,biotech位于波士顿、南加州都是首选,在这里科学技术、资本、人才、系统等都是最方便的。
本篇文章来源于微信公众号: 英勇向前